This article discusses a recent malignant hyperthermia (MH) case and addresses ways in which anesthesiologists can improve patient care in the management of the syndrome beyond the operating room. We introduce a concept called the "4C's of MH Management."
INTRODUCTION
As an anesthesia professional, you likely know that malignant hyperthermia (MH) strikes out of the blue and can rapidly lead to muscle rigidity, hyperthermia, elevated end tidal CO2, respiratory and metabolic acidosis, and if not treated promptly and specifically, it can result in death. You also may know that, despite MH being inherited in an autosomal dominant manner, there is a higher incidence in males than females and that the syndrome occurs at a higher rate in children than adults.1
Here we present a recent case of MH, give a historical perspective on the success anesthesia professionals have had in furthering our understanding of MH, and propose a future direction in which we may also play a critical role in caring for patients long after their acute episode—to ensure proper workup and understanding of post-episode sequelae.
MH CASE
An otherwise healthy 11-year-old girl presents to the hospital to have an incidentally discovered ovarian teratoma removed laparoscopically. She had no prior surgeries or encounters with anesthesia. Soon after induction of anesthesia with sevoflurane, paralysis with rocuronium, and intubation, she developed persistent tachycardia (heart rate 120 bpm from a baseline in the 80s), her temperature rose to 39.4°C, and her end-tidal CO2 (ETCO2) climbed to 110 mmHg. Muscle rigidity was also observed in the patient’s arms despite having received non-depolarizing neuromuscular blockade. As soon as these MH signs presented, help was called to assist in managing the crisis. The sevoflurane was discontinued, and Ryanodex® (dantrolene sodium, Eagle Pharmaceuticals, Inc., Woodcliff Lake, NJ, USA) was quickly administered. Within a few minutes, her heart rate, temperature, ETCO2, and musculoskeletal tone all improved. Other stabilization measures included starting a total intravenous anesthesia (TIVA), obtaining additional IV access and an arterial line, active cooling with administration of cold IV fluids, and aggressive hydration. Once stabilized, the surgery was quickly completed, and the patient transferred to the ICU. Dantrolene treatment was continued. Notably her first creatine kinase (CK) was abnormally high at 34,000 IU. The patient was extubated the following day. Over the next few days, she continued to improve, and her rhabdomyolysis resolved.
Over multiple visits in the days following the event, the anesthesia professionals learned that the patient had previously had episodes of heat intolerance and that her grandmother had an incidental finding of chronically elevated CK, but had not undergone a muscle biopsy. A comprehensive genetic panel testing 129 genes associated with rhabdomyolysis and metabolic myopathy was ordered along with consultation with a geneticist and neurologist. The genetic testing revealed an alteration in the ryanodine receptor (RYR1), confirming the diagnosis of MH. Interestingly, while she was found to have a RYR1 variant which is officially characterized as a variant of unknown significance (VUS) by the National Human Genome Institute and American College of Medical Genetics, it is considered a pathogenic variant by the European MH group, and has previously been associated with elevated CK levels, rhabdomyolysis, and weakness.2 She was ultimately discharged on postoperative day 5 with close follow-up with the medical genetics, neurology, and neuromuscular clinics.
MH: A SUCCESS STORY
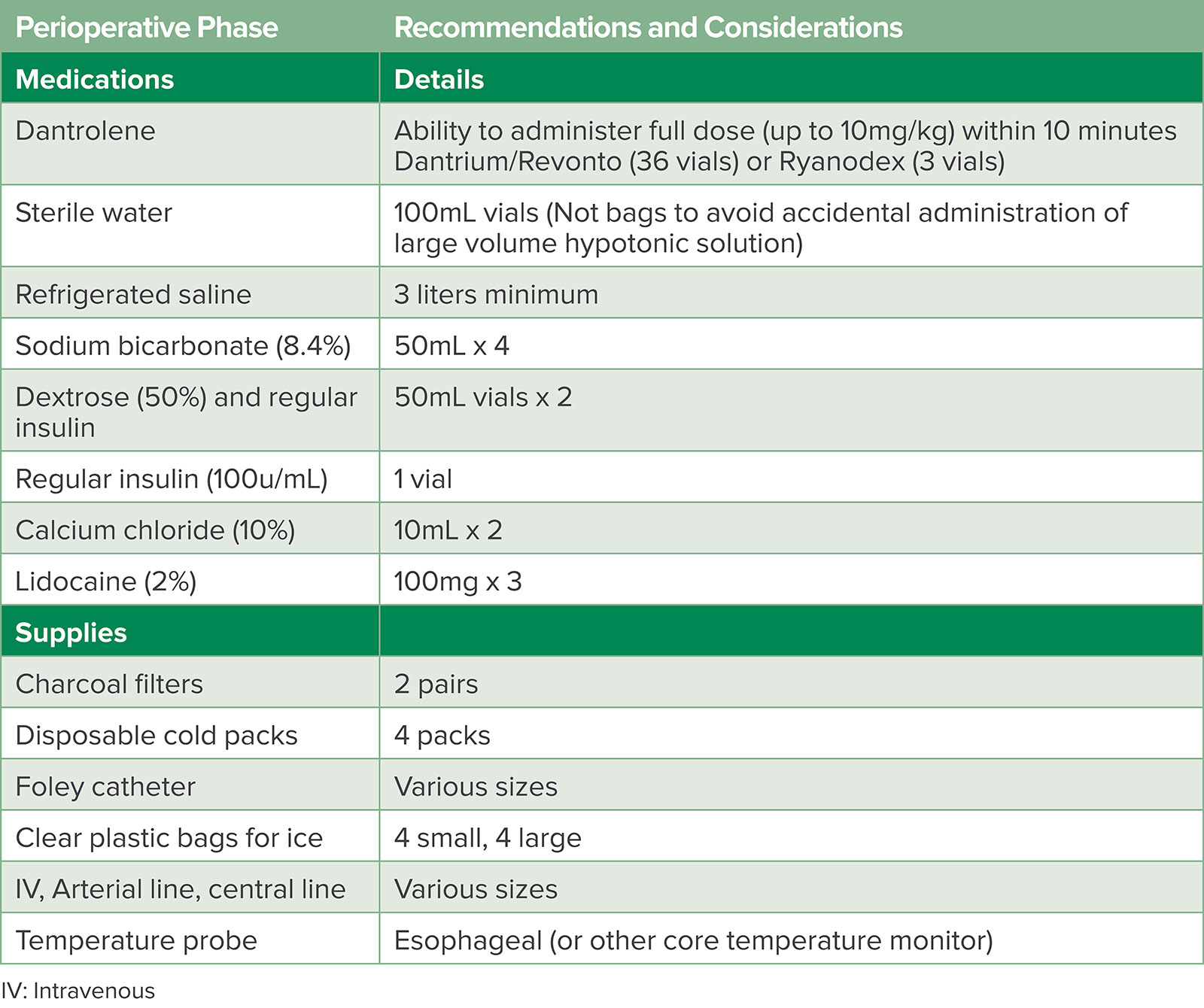
This story of a patient developing MH and being successfully diagnosed and treated reflects the accumulated knowledge from scientific contributions of scientists over many years and from all over the world. Since the first description of MH in Melbourne, Australia, in 1960 by Denborough and Lovell, much progress has been made in terms of defining the pathophysiology, discovering an effective therapeutic treatment, and disseminating information about MH to the anesthesia community.3 As a result, MH mortality has declined from 70% to as low as about 10% in countries prepared for the syndrome with ready access to dantrolene and other supplies (Table 1),4 a coordinated approach to diagnosis and management including ICU observation, and post-episode coordination with family members of the person who experienced MH.
Table 1: MH Cart Medication and Supplies.
FUTURE DIRECTIONS
As in much of science, solving one problem often leads to more questions. In addressing these questions, it has become apparent that we should not think of MH as an episodic, idiosyncratic disorder, but as a pharmacogenetic disorder and an inherited myopathy with consequences that may be far removed from the anesthetic experience.
First, what do we know about the implications of RYR1 mutations outside of MH? Further study is needed to understand how RYR1 associated conditions (such as statin-induced myopathy, exertional rhabdomyolysis, heat stroke, and chronically elevated CK syndromes) relate to MH susceptibility (MHS).5 There is also a need to understand the role and efficacy of dantrolene for the treatment of these other syndromes.
Second, after an MH episode, how should we confirm a diagnosis of MHS? Advances in genetic testing to assess MHS have led genetics to essentially replace the tried-and-true muscle biopsy caffeine halothane contracture test in much of the world. The European MH society formally changed their recommendation to use genetics as the first line test in MHS diagnosis in 2015 and most MH experts around the world have followed this lead.6 In the US, there are only two centers (located in Minnesota and North Carolina) and the testing can cost up to $20,000 USD, which is generally not paid for by insurance companies. On the other hand, the cost of genetic testing has declined over 99% since the first sequencing of the human genome and is usually reimbursed by insurance companies. Specific costs vary by the type of test selected. A panel test for the three genes associated with MHS (RYR1, CACNA1S, and STAC3) costs less than $500 USD,7 versus a more comprehensive metabolic and myopathy panel which costs around $1,500 USD.8
Third, what is the role of the anesthesia professional in the care of MHS patients after their episode, and how should they interact with other providers such as geneticists and neurologists? Whose responsibility is it to advise family members on the significance of a pathogenic DNA variant predisposing to MH? Specialists who treat patients who have experienced an MH episode can help them to understand whether they are expected to make a full recovery, but they may be less able to address longer lasting questions such as if such patients are predisposed to rhabdomyolysis or muscle weakness, and for how long. A method of flagging an MHS diagnosis, including specific genetic variant, in the electronic medical record should be ensured. Artificial intelligence or third-party services that can process data across genetic testing platforms and improve communication of this critical diagnosis to practitioners can make care safer for MHS patients. Though the incidence of acute MH in MHS patients exposed to triggering agents is unknown and difficult to estimate, all MHS patients should be treated with a “clean” nontriggering anesthesia technique.9-11 This includes avoidance of succinylcholine and volatile anesthetics, preparation of the anesthesia machine by flushing the circuit and ventilator with high flows over the manufacturer recommended amount of time or insertion of activated charcoal filters in the breathing circuit, use of fresh CO2 absorbent, and taping off vaporizers to prevent accidental use. Intraoperative monitors should include EKG, pulse oximetry, blood pressure, core body temperature, and capnography for general anesthesia.12
Finally, MH has been reported from countries all over the world and there is no known ethnic propensity to MHS. A broader public health concern is how do resource-limited countries balance the cost of a life-saving drug dantrolene for an infrequent disorder, versus the need for expenditures on more frequently occurring disorders? A report from China in the February 2024 issue of Anesthesiology reported mortality from MH over 50% in areas without dantrolene.13 While it has been questioned in the US as to whether ambulatory facilities who do not routinely use volatile agents and reserve succinylcholine for emergency airway management need to stock dantrolene, there is evidence to show that stocking dantrolene is both cost-effective and optimal for safe patient care where triggering agents may possibly be used.14-17
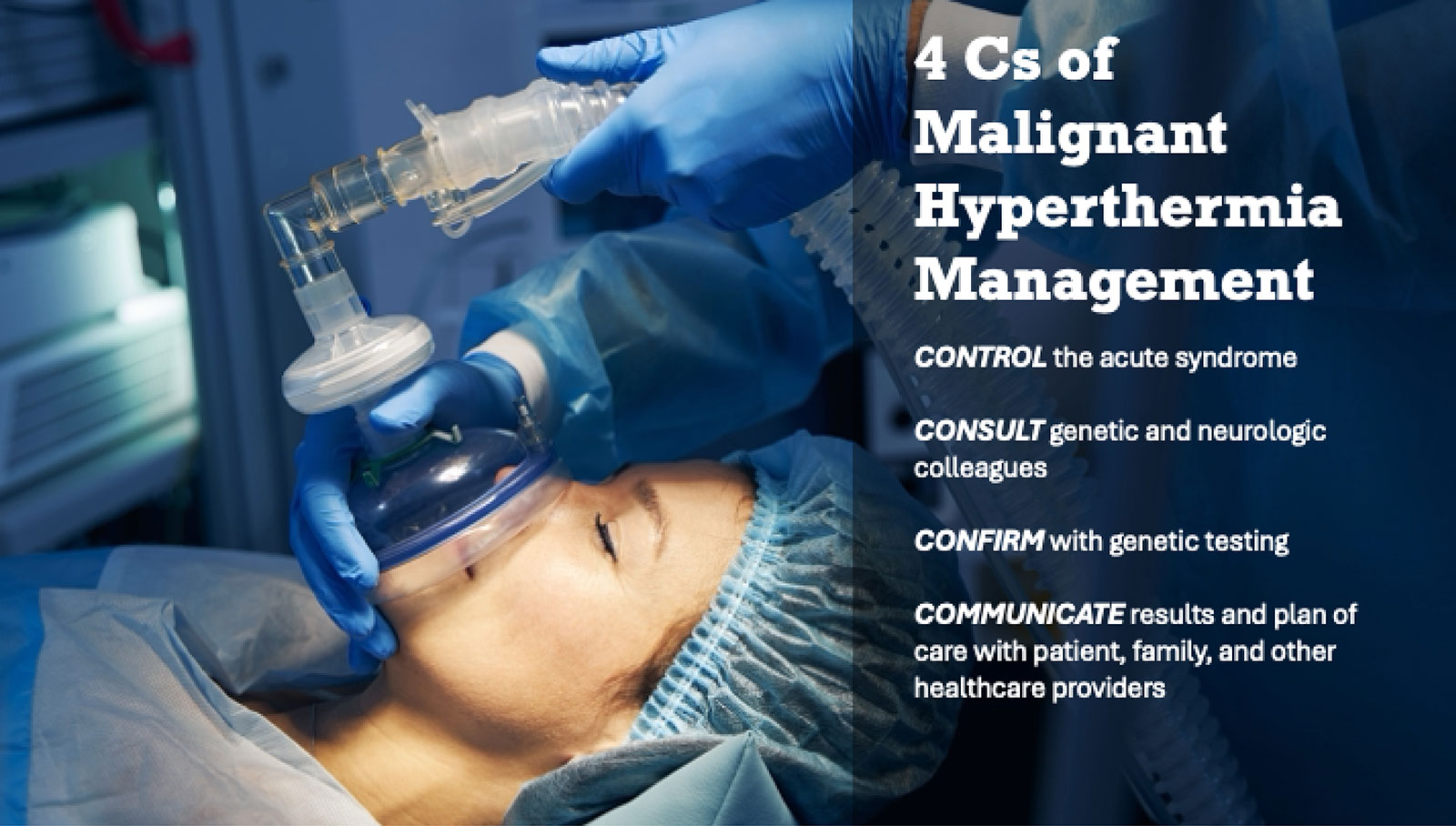
THE FOUR C’S OF MH MANAGEMENT
In our opinion it is time to broaden our perspective on the MH syndrome to regard the problem as not unique to anesthesiology and surgery. We need to expand patient care goals beyond the confines of managing the acute MH episode, to include appropriate follow-up and assessment of other potentially associated skeletal-muscle-related conditions. A multidisciplinary approach to the diagnosis, treatment, and genetic counseling for patients and families is of utmost importance. Therefore, we propose the 4C’s of MH Management:
- Control the acute syndrome
- Consult genetic and neurologic colleagues
- Confirm with genetic testing
- Communicate the results and plans for care with the patient, family, and other health care providers.
NEXT STEPS
It is within our scope of practice as anesthesia professionals to take these actions to improve the care of MHS patients going forward. The enlightened management of MH fits with the goals of the APSF and other patient safety organizations. Next steps include: 1) revision of MHS diagnostic testing recommendations by the Malignant Hyperthermia Association of the United States (MHAUS) to include genetic testing after all suspected MH cases, 2) creation of a system to connect anesthesia professionals who manage MH to clinical geneticists, genetic testing platforms, and neurologists familiar with MH, 3) assurance of communication of an MHS diagnosis in electronic medical records, and 4) dissemination of MH education globally, including advocating for stocking dantrolene in all centers where triggering agents may be used.
Henry Rosenberg, MD, is a president emeritus of the Malignant Hyperthermia Association of the United States (MHAUS).
Anjan Saha, MD, is a clinical anesthesia year 3 (CA3) resident (Apgar Scholars’ Program) at Columbia University, New York, NY.
Carla D. Zingariello, DO, is an assistant professor of pediatrics at the University of Florida College of Medicine, Gainesville, FL.
Sandra Natalia Gonzalez, MD, FAAP, is an assistant professor of anesthesiology at the University of Florida College of Medicine, Gainesville, FL.
Teeda Pinyavat, MD, is an associate professor of anesthesiology at Columbia University, New York, NY.
Henry Rosenberg, MD serves as emeritus president of MHAUS and, member of the Board of Directors of MHAUS
Anjan Saha, MD is an equity holder in a start-up company called, Deoxylytics. Deoxylytics is a pharmacogenomics firm.
Carla Zingariello, DO is a principal investigator for a clinical trial with ML Bio and has served as a consultant for ML Bio.
Sandra Natalia Gonzalez, MD, has no conflicts of interest.
Teeda Pinyavat, MD, is a member of the MHAUS Board of Directors.
REFERENCES
- Rosenberg H, Pollock N, Schiemann A, et al. Malignant hyperthermia: a review. Orphanet J Rare Dis. 2015;10:93. PMID: 26238698
- Witting N, Laforêt P, Voermans NC, et al. Phenotype and genotype of muscle ryanodine receptor rhabdomyolysis-myalgia syndrome. Acta Neurol Scand. 2018;137:452–461. PMID: 29635721
- Denborough MA. Malignant hyperthermia. 1962. Anesthesiology. 2008;108:156–157.
- Malignant Hyperthermia Association of the United States (MHAUS) Professional Advisory Council. “What should be on an MH cart?” MHAUS.org. https://www.mhaus.org/healthcare-professionals/be-prepared/what-should-be-on-an-mh-cart/. Accessed July 20, 2024.
- Fiszer D, Shaw MA, Fisher NA, et al. Next-generation sequencing of RYR1 and CACNA1S in malignant hyperthermia and exertional heat illness. Anesthesiology. 2015;122:1033–1046. PMID: 25658027
- Hopkins PM, Rüffert H, Snoeck MM, et al. on behalf of European Malignant Hyperthermia Group. European Malignant Hyperthermia Group guidelines for investigation of malignant hyperthermia susceptibility. Br J Anaesth. 2015;115:531–539. PMID: 26188342
- “Invitae Malignant Hyperthermia Susceptibility Panel.” Invitae. https://www.invitae.com/us/providers/test-catalog/test-03285. Accessed July 20, 2024.
- “Metabolic Myopathy and Rhabdomyolysis Panel.” Blueprint Genetics. https://blueprintgenetics.com/tests/panels/neurology/metabolic-myopathy-and-rhabdomyolysis-panel/. Accessed July 20, 2024.
- Ibarra Moreno CA, Hu S, Kraeva N, et al. An assessment of penetrance and clinical expression of malignant hyperthermia in individuals carrying diagnostic ryanodine receptor 1 gene mutations. Anesthesiology. 2019;131:983–991. PMID: 31206373
- Shaw MA, Hopkins PM; Mission impossible or mission futile?: estimating penetrance for malignant hyperthermia. Anesthesiology. 2019;131:957–959. PMID: 31335544
- Yu KD, Betts MN, Urban GM, et al. Evaluation of malignant hyperthermia features in patients with pathogenic or likely pathogenic RYR1 variants disclosed through a population genomic screening program. Anesthesiology. 2024; 140:52–61. PMID: 37787745
- Rüffert H, Bastian B, Bendixen D, et al. European Malignant Hyperthermia Group. Consensus guidelines on perioperative management of malignant hyperthermia suspected or susceptible patients from the European Malignant Hyperthermia Group. Br J Anaesth. 2021;126:120–130. PMID: 33131754
- Yu H, Tan L, Deng X. Improving dantrolene mobilization in regions with limited availability. Anesthesiology. 20241;140:1201–1202. PMID: 38329334
- Joshi GP, Desai MS, Gayer S, Vila H Jr. Society for Ambulatory Anesthesia (SAMBA). Succinylcholine for emergency airway rescue in class B ambulatory facilities: the Society for Ambulatory Anesthesia position statement. Anesth Analg. 2017;124:1447–1449. PMID: 27984222
- Litman RS, Smith VI, Larach MG, et al. Consensus statement of the Malignant Hyperthermia Association of the United States on unresolved clinical questions concerning the management of patients with malignant hyperthermia. Anesth Analg. 2019;128:652–659. PMID: 30768455
- Larach MG, Klumpner TT, Brandom BW, et al.; Multicenter Perioperative Outcomes Group. Succinylcholine use and dantrolene availability for malignant hyperthermia treatment: database analyses and systematic review. Anesthesiology. 2019;130:41–54. PMID: 30550426
- Aderibigbe T, Lang BH, Rosenberg H, et al. Cost-effectiveness analysis of stocking dantrolene in ambulatory surgery centers for the treatment of malignant hyperthermia. Anesthesiology. 2014;120:1333–1338. PMID: 24714119
