Introduction
Following the great fire of London in 1666, the first automated detector and threshold fire alarm was invented. This alarm was comprised of a string that stretched through each room of a house, and then extended to the basement where it was connected to a weight suspended over a gong. In theory, a fire in this “threshold monitored” home would burn through the string and trigger the alarm, resulting in a “better late than never” arousal of its occupants. Today, hospital care-givers and their patients still rely on this simple threshold alarm model, substituting threshold values of SPO2, RR, heart rate, and etCO2 as clinical surrogates for the string. Unfortunately, clinical trials1,2 and a recent comprehensive research review3 suggest that these threshold monitors, like the string, are not as effective as their designers first believed.
With our evolving recognition of the weakness single thresholds provide, variations on the threshold alarm method, such as the modified early warning score (MEWS), have been introduced. The MEWS system generates numeric scores from a range of threshold breaches and then adds these scores to produce a super “fusion” threshold. While MEWS may be an improvement, as we will see, it suffers from significant risk inducing anomalies inevitable whenever simple addition is used to quantify complex pathophysiologic processes.
One reason threshold monitors and MEWS may not be as effective as expected on hospital general care floors is that patients often die unexpectedly by progression through a range of 3 common, but distinctly different dynamic patterns of instability. We call these “Patterns of Unexpected Hospital Death” (PUHD) (Table 1). While these death patterns are not overly complex, they cannot be detected early by any single or multi-parameter threshold breach.

The Common Patterns of Unexpected Hospital Death (PUHD)
Type I Pattern of Unexpected Hospital Death (e.g., Sepsis, CHF, PE)
This pattern reflects a clinically evolving process associated with microcirculatory failure induced by such common conditions as CHF, sepsis, and pulmonary embolism. The pattern generally begins with subtle hyperventilation and a persistent respiratory alkalosis (RA) despite subsequent progressive increases in anion gap and lactic acid levels. This stage occurs well before the development of dominate metabolic acidosis (MA), which is usually associated with its late and terminal stages. These progressive pattern phases (initial isolated RA followed by mixed RA and MA, followed by dominate MA) comprise the typical progression of Type I PUHD, and are shown in Figure 1.
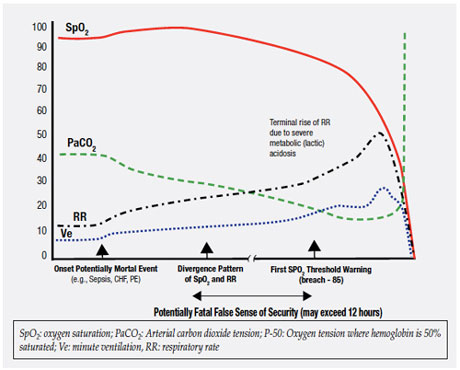
Figure 1. Type I Pattern of Unexpected Hospital Death (e.g., Sepsis, CHF).
Unfortunately, the very high respiratory rate thresholds (above 30/min) commonly used to trigger rapid response team activations,5,6 occur most often in non-survivors7 with no evidence showing they are breached early in sepsis or other conditions producing the Type I PUHD. Very high respiratory rates (above 30/min), like high lactate levels,8 are likely to assist detection when severe metabolic acidosis, a late Type I PUHD manifestation, enters the picture. These are best considered markers of severity and diagnostic delay9 rather than useful warnings for early disease.
Eventually microcirculatory failure in the lungs causes a fall in PaO2,10 but hyperventilation can perpetuate SPO2 values well above 90% regardless of a falling PaO2 because of respiratory alkalosis.11 It’s precisely these early, compensatory physiologic changes, and the oximetry patterns from Type I PUHD, which can fool clinicians into mistakenly believing these patients aren’t in trouble.
The failure of single thresholds has led to the development of reliance on multiple perturbed parameters combined to generate a Modified Early Warning Score (MEWS). However, reliance on the sum of threshold perturbations of multiple parameters presents unique problems. In just one example, a heart patient receiving a beta blocker may require a higher respiratory rate to achieve a threshold MEWS score than a patient without heart disease. These types of anomalies illustrate the weakness of oversimplified and arbitrary scoring like MEWS.
To summarize, this unique Type I process starts with a rising minute ventilation and a falling PaCO2, then a late slow fall in SPO2 and a more rapid rise in minute ventilation (and at this point a severe rise in respiratory rate and marked additional fall in PaCO2), followed then by a rapid drop in SPO2 (often only now passing through the SPO2 alarm threshold). If supplemental oxygen is provided, the SPO2 can remain stable even closer to the death point, prolonging the false sense of security. Threshold breaches of RR, SPO2, or the MEWS are generally late and unpredictable markers of the Type I pattern.
Type II Pattern of Unexpected Hospital Death (CO2 Narcosis)
Since the 1950s,12 nurses and physicians in training have learned that narcotics produce death through a singular path involving progressive hypoventilation. Perceived as a deteriorating, self-propagating process, both the narcotics and a rising PaCO2 contribute to the central depression of ventilatory drive, ultimately leading to “CO2 Narcosis” severe enough to bring on respiratory arrest. As hypoventilation progresses, supplemental low flow oxygen can hide it entirely from the pulse oximeter until very late,13-15 just as it does with Type I PUHD.
Classic cases of this are seen in accidental narcotic overdose, and those patients with hypoventilation syndromes, such as adult patients with congenital central hypoventilation syndrome, e.g., PHO2XB mutations.16
In summary, (as illustrated in Figure 2) the Type II PUHD comprises first a progressive fall in minute ventilation due to declines in tidal volume and/or respiratory rate, both unpredictably variable. This induces a progressive rise in PaCO2 with the patient exhibiting progressively higher sedation scores to the point of stupor and death. Patients provided with supplemental oxygen can maintain SPO2 values in the 90-100% range until very late.
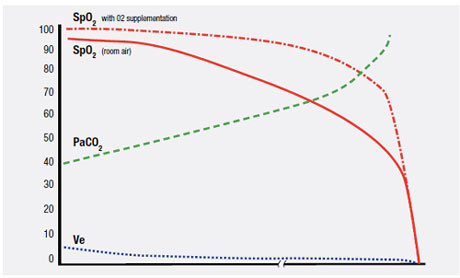
Figure 2. Type II Pattern of Unexpected Hospital Death (CO2 Narcosis).
Type III Pattern of Unexpected Hospital Death (Sleep Apnea)
Having just discussed the prevailing belief held for decades (and still being taught in Medical Schools) on the cause of respiratory failure and death induced by narcotics and sedatives, we’re now ready to unsettle any certainty and comfort this simplistic belief might provide. A “stand alone” Type II concept has fomented the widely held perception that sedation scores combined with threshold alarms from pulse oximeters and/or capnometry can reliably provide early detection.
Back in 2002, Lofsky17 described a cluster of unexpected hospital deaths involving patients with risk factors for obstructive sleep apnea. These patients died in bed in spite of acceptable dosing of narcotics. Surprisingly, they all shared a unique clinical course that started with being alert, then sleeping, and then dying within brief timelines. We now know that sleep apnea with arousal failure produces a distinct pattern during sleep, which we’ve named the Type III PUHD. It differs from our classic Type II CO2 narcosis process, in that it occurs only during sleep and may not be associated with prior elevated sedation scores. When awake, patients with profound Type III arousal failure may exhibit no pathognomonic symptoms or signs, or show evidence of any “awake” sedation. In other words, patients with arousal failure are orphaned, remaining completely concealed within our typical pre and postoperative populations. As shown in Figure 3, the sentinel instability component of Type III PUHD is induced by sleep apnea in the presence of arousal failure.
This Type III pattern architecture is comprised of repetitive reductions in airflow and SPO2 from sleep related cycling collapses of the upper airway.18,19 This cycling shown in Figure 4, with initial collapsing and then reopening of the upper airway, produces a typical and very distinctive pattern of signal clusters that is reliably acquired through high resolution pulse oximetry.
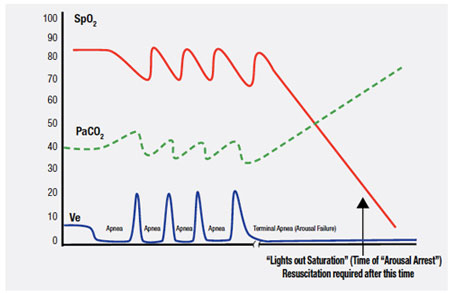
Figure 3. Type III Pattern of Unexpected Hospital Death (Sleep Apnea with Arousal Failure).
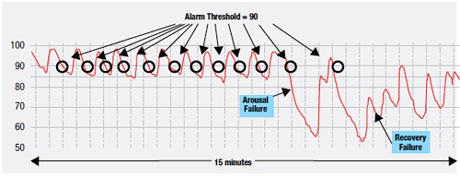
Figure 4. Type III Pattern: Note the Potential for Alarm Fatigue Preceding Arousal Failure.
Obstructive sleep apnea can be best understood as a condition where during sleep, one’s upper airway collapses and is held closed by vigorous but ineffective respiratory effort (much like trying to suck on a collapsed cellophane straw). Each apnea in a repetitive sequence of cyclic apneas is generally terminated by a micro-arousal. The arousal then causes brief “overshoot” hyperventilation that drives the PaCO2 below normal. This drop in PaCO2 triggers a fall in central ventilation drive and upper airway tone. Since the upper airway is already unstable it collapses again, causing the cycle to reenter and self-propagate, producing its sentinel pattern of repetitive reductions in airflow and SPO2.18 Narcotics,20-22 spinal anesthesia,23 sedatives,24 and cycling hypoxemia25 can increase the arousal threshold (cause arousal delay), and then respiratory arrest can occur from complete arousal failure (arousal arrest).26,27 Once this occurs, if no intervention is provided immediately, a Type III death will follow suddenly during sleep without warning due to precipitous hypoxemia, and most often without much progressive PaCO2 elevation because of insufficient time for hypercarbia to develop.
It has been postulated that chronic arousal failure may develop as a function of neural plasticity in response to repetitive exposures to rapid declines in oxygen saturation over many years. As the central arousal system adjusts its response, the arousal itself can become progressively more delayed (much as it would to intermittent loud sounds after years of sleep exposure to the passing of nearby trains). By the time the patient, exposed to many years of repetitive desaturations every night, arrives for surgery, the arousal failure may have unknowingly progressed to profoundly low pre-op levels.
One reason arousal delay becomes so critical is that SPO2 is able to fall at very rapid rates during apnea. Many physicians accustomed to witnessing preoxygenated apnea lack a full appreciation for the extremely early and very steep desaturation slopes seen in recumbent, obese patients with apnea. In fact, since postoperative functional residual capacity does not have definable lower limits, oxygen desaturation rates may in some cases exceed 1.5% per second with SPO2 falling to critical values with no time for contemporaneous hypercarbia to develop.28 Occasionally a patient’s arterial oxygen saturation falls to a point where the brain no longer receives sufficient oxygen for central arousal to occur.21,26,27 This is called the “Lights Out Saturation” (LOS) and happens because the human brain is incapable of generating sufficient anaerobic metabolism and depends on a continuous supply of oxygen to support arousal. If arterial oxygen saturations fall below this critical value where the hemoglobin molecule simply cannot release sufficient oxygen to the brain, EEG slowing occurs promptly and arousal becomes totally suppressed: the “lights go out.”
Once the LOS is breached, airway reopening without resuscitation isn’t to be expected. The body remains alive and continues to burn glucose and fat, and the heart pumps ever mounting CO2 stores through an anoxic body. If the patient is discovered now and resuscitation initiated, the immediately drawn blood gas will show the PaCO2 to be quite high, disguising this incident as a Type II event.
In summary, if unrecognized sleep apnea with its unique state of arousal dependent survival exists, the cycling SPO2 signals can provide sentinel markers for both cyclical apnea and arousal failure. Unknowing administration of narcotics and/or sedatives to patients with preexisting arousal failure can delay an already failing arousal to the point of arousal arrest.
Discussion
Like the London string, the primary limitations of threshold monitors are due to their oversimplified design. If there was only one pattern of a house fire, or one pattern of unexpected respiratory instability, it might be possible to find a “best” string position in the house, and a best clinical threshold in the hospital. However, there are 3 common patterns of unexpected hospital death, all at counter purpose to one another regarding their detection, effects on physiology, and potential for alarm fatigue. Optimize a threshold to reduce alarm fatigue for one pattern and you inadvertently place patients suffering from the other patterns at risk for greater delays. Thresholds which appear effective in one population with a high grouping of one pattern may fail in another population with a different distribution. For this reason, alarm research studies must identify the distribution of patterns rendering the alarms before any conclusions can be drawn. Finally, “beeps” and/or MEWS that do not tell the health care worker which death pattern is evolving, and how far advanced the death pattern is, are easy to ignore and provide too little information for action.
However old, threshold devices still do have benefits, and these are the only devices presently available to protect our patients. Change to more advanced alarm processing technology will take time. One immediate solution is to expedite the development of a training course (analogous to the advance cardiac life support course) to certify all health care workers using patient monitors. This training would include modules designed to teach the PUHDs, the technical and pathophysiologic causes of alarm fatigue, and the benefits/limitations of monitoring and sedation scoring in relation to each distinct death pattern.
Formal training would also help prevent delay and death due to threshold based “technical trivialities” such as a patient’s MEWS changing too late from a score of 3 to 4, or the generation of alarm fatigue by a death pattern which produces many early “threshold breaches” before an actual death event occurs, or a failure to alarm at all from the threshold monitoring of an unappreciated, compensated parameter.
An understanding of the relational and conformational complexity of the PUHDs also argues strongly for computational transparency of all alarm processors, which simply means that the original clinical data set, the processed data set, and the basis for outputs as a function of the processing are exposed (or readily exposable) in real time at the bedside by the clinical care-givers managing patients. Physicians should take charge of this process.
Finally, a major focus on improving patient monitors and clinical trials is required. Patients are dying in hospitals with smart phones in their pockets that can identify a song just by “listening” to it, while the monitors they are connected to are not smart enough to identify even one pattern of unexpected death.
Conclusion
There are 3 common fundamental pathophysiologic patterns of unexpected hospital death. These patterns are too complex for early detection by any unifying numeric threshold or summation score. Furthermore, alarms responsive to simple fragments of patterns (e.g., thresholds or trends) rather than the patterns themselves have the potential to induce alarm inflation. Those using or designing patient monitors should receive formal training relevant to the patterns of unexpected hospital death. Clinical trials on alarms should identify the distribution of the patterns that generated them. In addition, new methods and technologies which detect, identify, quantify and track the actual patterns of unexpected hospital death should be investigated. It’s time to cut the string.
Department of Anesthesiology and Perioperative Care, Newport Beach, CA. and a Clinical Professor, Department of Anesthesiology, David Geffen School of Medicine, UCLA.
Dr. Lynn is a pulmonary and critical care physician and serves as executive director of the Sleep and Breathing Research Institute in Columbus Ohio. He also serves on the FDA standards committee for pulse oximetry monitoring.
Disclosure: Dr. Curry has nothing to disclose. Dr. Lynn holds patents and receives royalties relating to inventions in the field of patient monitoring and pattern detection.
References
- Chan PS, Khalid A, Longmore LS, et al. Hospital-wide code rates and mortality before and after implementation of a rapid response team. JAMA 2008;300:2506-13.
- Rothschild JM, Woolf S, Finn KM, et al. A controlled trial of a rapid response system in an academic medical center. Jt Comm J Qual Patient Saf 2008;34:417-25, 365.
- Chan PS, Jain R, Nallmothu BK, et al. Rapid response teams: A systematic review and meta-analysis. Arch Intern Med 2010;170:18-26.
- Ghanem-Zoubi NO, Vardi M, Laor A, et al. Assessment of disease-severity scoring systems for patients with sepsis in general internal medicine departments. Crit Care 2011;15:R95.
- Hravnak M, Edwards L, Clontz A, et al. Defining the incidence of cardiorespiratory instability in patients in step-down units using an electronic integrated monitoring system. Arch Intern Med 2008;168:1300-8.
- Alian AA, Rafferty T. Evaluation of rapid response team flag-alert parameters. Available at: http://www.cardiopulmonarycorp.com/pdf/RapidResponseAlertParameters.pdf. Accessed August 11, 2011.
- Bossink AW, Groeneveld J, Hack CE, et al. Prediction of mortality in febrile medical patients: How useful are systemic inflammatory response syndrome and sepsis criteria? Chest 1998;113:1533-41.
- Mikkelsen ME, Miltiades AN, Gaieski DF, et al. Serum lactate is associated with mortality in severe sepsis independent of organ failure and shock. Crit Care Med 2009;37:1670-7.
- Marini C, Di Ricco G, Formichi B, et al. Arterial base deficit in pulmonary embolism is an index of severity and diagnostic delay. Intern Emerg Med 2010;5:235-43.
- Greer R. The temporal evolution of acute respiratory distress syndrome following shock. Eur J Anaesthesiol 2010;27:226-32.
- Siggaard-Andersen O, Garby L. The Bohr effect and the Haldane effect. Scand J Clin Lab Invest 1973;31:1-8.
- Seiker HO, Hickam JB. Carbon dioxide intoxication: the clinical syndrome, its etiology and management with particular reference to the use of mechanical respirators. Medicine (Baltimore) 1956;35:389-423.
- Ayas N, Bergstrom LR, Schwab TR, et al. Unrecognized severe postoperative hypercapnia: a case of apneic oxygenation. Mayo Clin Proc 1998;73:51-4.
- Fu ES, Downs JB, Schweiger JW, et al. Supplemental oxygen impairs detection of hypoventilation by pulse oximetry. Chest 2004;126:1552-8.
- Downs JB. Has oxygen administration delayed appropriate respiratory care? Fallacies regarding oxygen therapy. Respir Care 2003;48:611-20.
- Antic NA, Malow BA, Lange N, et al. PHOX2B mutation-confirmed congenital central hypoventilation syndrome: presentation in adulthood. Am J Respir Crit Care Med 2006;174:923-7.
- Lofsky A. Sleep apnea and narcotic postoperative pain medication: A morbidity and mortality risk. APSF Newsletter 2002;17:24-5.
- Lynn LA. Interpretive oximetry: future directions for diagnostic applications of the SpO2 time-series. Anesth Analg 2002;94(1 Suppl):S84-8.
- Dempsey JA, Veasey SC, Morgan BJ, et al. Pathophysiology of sleep apnea. Physiol Rev 2010;90:47-112.
- Hajiha M, DuBord MA, Liu H, et al. Opioid receptor mechanisms at the hypoglossal motor pool and effects on tongue muscle activity in vivo. J Physiol 2009;587:2677-92.
- White DP. Opioid-induced suppression of genioglossal muscle activity: is it clinically important? J Physiol 2009;587:3421-2.
- VanDercar DH, Martinez AP, De Lisser EA. Sleep apnea syndromes: a potential contraindication for patient-controlled analgesia. Anesthesiology 1991;74:623-4.
- Wieczorek PM, Carli F. Obstructive sleep apnea uncovered after high spinal anesthesia: a case report. Can J Anaesth 2005;52:761-4.
- Berry RB, Kouchi K, Bower J, et al. Triazolam in patients with obstructive sleep apnea. Am J Respir Crit Care Med 1995;151:450-4.
- Johnston RV, Grant DA, Wilkinson MH, et al. Repetitive hypoxia rapidly depresses cardio-respiratory responses during active sleep but not quiet sleep in the newborn lamb. J Physiol 1999;519:571-9.
- Khoo SM, Mukherjee JJ, Phua J, et al. Obstructive sleep apnea presenting as recurrent cardiopulmonary arrest. Sleep Breath 2009;13:89-92.
- Dyken ME, Yamada T, Glenn CL, et al. Obstructive sleep apnea associated with cerebral hypoxemia and death. Neurology 2004;62:491-3.
- Wilkinson MH, Berger PJ, Blanch N, et al. Effect of venous oxygenation on arterial desaturation rate during repetitive apneas in lambs. Respir Physiol 1995;101:321-31.
